Genetic regulation of cell type–specific chromatin accessibility shapes brain disease etiology
Abstract
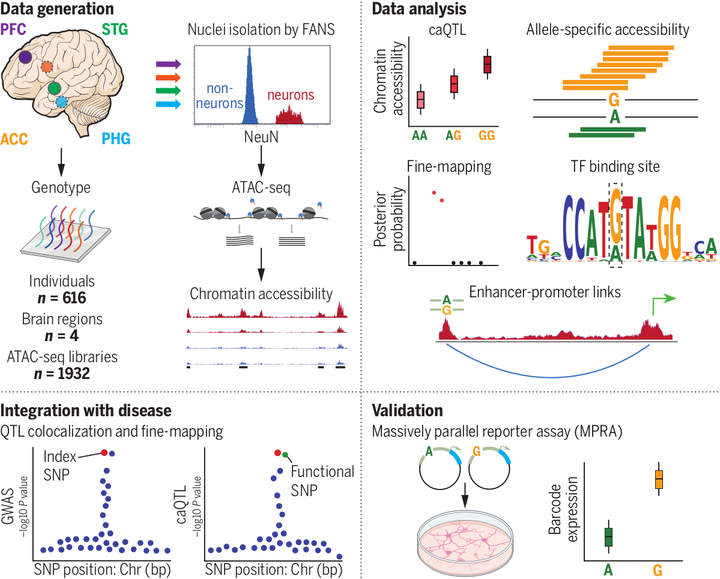
INTRODUCTION. Genome-wide association studies (GWASs) have led to the identification of hundreds of genetic loci that are associated with increased risk for a variety of brain diseases, including schizophrenia, Alzheimer’s disease, bipolar disorder, and major depressive disorder. However, identifying molecular mechanisms that underlie GWAS findings remains challenging because of linkage disequilibrium between genetic variants. Additionally, many risk variants are located in noncoding regions of the genome that are enriched in regulatory elements, which suggests that these variants affect gene expression rather than protein structure and function. Examining expression quantitative trait loci (eQTLs), especially cell type–specific eQTLs, has recently identified genetic regulatory signals that are shared between gene expression and brain disease traits. Yet our understanding of cell type–specific regulatory processes that drive variation in molecular traits mediating disease risk remains limited – RATIONALE. Regulatory sequences, including promoters, enhancers, insulators, and transcriptional silencers, are enriched in open chromatin regions (OCRs). Importantly, genetic variants that influence chromatin accessibility can, in turn, either activate or suppress gene expression by disrupting the function of these regulatory elements. Because chromatin status is directly connected to the regulation of transcriptional activity, the presence of colocalized chromatin accessibility QTLs (caQTLs) and eQTL signals within a given locus can reveal functional regulatory elements, connecting risk variants to causative genes and disease processes. Finding disease-relevant regulatory elements, especially those with cell type–specific effects, may provide the means to identify therapeutic targets for a range of neuropsychiatric and neurodegenerative disorders – RESULTS. To investigate the genetic regulation of chromatin accessibility in the brain, as well as its impact on disease, we analyzed 1932 cell type–specific ATAC-seq (assay for transposase-accessible chromatin with sequencing) libraries consisting of neurons and non-neurons isolated from four functionally distinct brain regions of 616 human postmortem brains. Our study identified 34,539 OCRs with caQTLs and shows that the genetic control of chromatin accessibility displays a high degree of cell-type specificity. Using statistical fine-mapping, eQTL and caQTL colocalization, and allele-specific chromatin accessibility, we identified potential molecular mechanisms that mediate the effects of disease risk variants. The integration of caQTL and eQTL results with GWAS results from six brain diseases identified 72 genes and 92 OCRs that mediate disease risk. By testing the functional impact of 19,893 potential causal variants in human induced pluripotent stem cell–derived excitatory neurons, using a massively parallel reporter assay (MPRA), we identified 476 variants with allelic effects. Genome-wide, we found that annotating variants on the basis of neuronal chromatin accessibility, as well as statistical fine-mapping from neuronal caQTL and eQTLs from brain homogenate, can be used to predict the allelic fold change in the MPRA. The MPRA identified an allelic effect in the variant rs3764512, which is predicted to increase local chromatin accessibility in neurons, increase expression of RAB27B, and increase risk for major depressive disorder.